Rabdomiosarcoma (RMS)
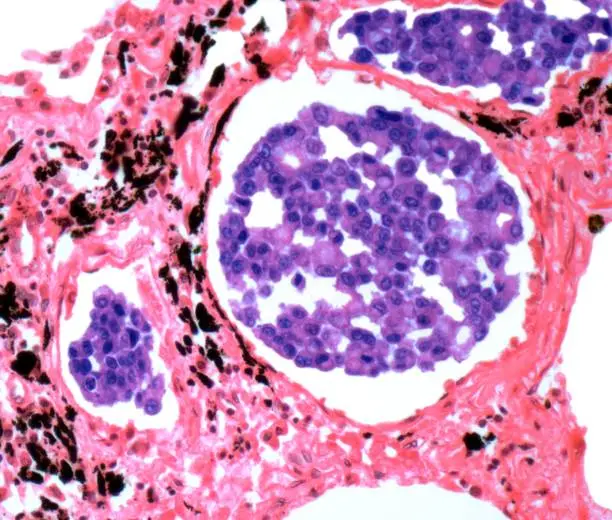
Il rabdomiosarcoma (RMS) è un tumore maligno raro e aggressivo che origina dalle cellule progenitrici del muscolo scheletrico e colpisce più comunemente bambini e adolescenti. Rappresenta il sarcoma dei tessuti molli più frequente nella popolazione pediatrica e può insorgere in diverse parti del corpo, tra cui la regione della testa e del collo, il tratto genitourinario e gli arti. Esistono due principali sottotipi istologici: il rabdomiosarcoma embrionale, più comune nei bambini più piccoli e con una prognosi relativamente migliore, e il rabdomiosarcoma alveolare, che si osserva tipicamente negli adolescenti ed è associato a un decorso clinico più aggressivo. La causa esatta del RMS non è completamente compresa, ma mutazioni genetiche e traslocazioni cromosomiche, come le fusioni PAX3-FOXO1 o PAX7-FOXO1, sono state implicate nel suo sviluppo. La diagnosi si basa su studi di imaging ed è confermata da biopsia e analisi immunoistochimica. Il trattamento prevede in genere una combinazione di chirurgia, chemioterapia e radioterapia, personalizzata in base alla sede, allo stadio e al sottotipo del tumore. Nonostante i progressi nel trattamento multimodale, la prognosi del rabdomiosarcoma metastatico o ricorrente rimane infausta, il che sottolinea la necessità di una ricerca continua e di innovazioni terapeutiche.